Genomalysis User Guide
PDF Version (250 KB).
Table of Contents
- A Note
- Introduction
- Graphical Layout
- Filter Algorithms
- Notes on Algorithm Use
- Sequence Length Filter
- Regex Filter
- Transmembrane Prediction Filter
- Secretion Signal Filters
- Clustal Omega Filter
- A Primer on Regex
- Fasta Files
- Algorithms for DNA and Proteins Sequences
Filter Algorithms
Notes on Algorithm Use. All filter algorithms available in Genomalysis are queued for execution by double clicking on them in the available filters dialog shown in the Filter Sequences tab of the main Genomalysis window, as shown above in the Graphical Layout section. When an available filter is double clicked, it will cause a filter instance of that filter type to appear in the filter instances dialog. Double clicking a second available filter will cause a filter instance of the second filter type to appear below the first in the filter instances dialog. This can be repeated as many times as necessary to cover all parameters that the user wishes to filter for in a sequence search. Because all filters are queued and executed the same way, only the specifics of each algorithm's configuration options will be discussed here.Filter instances are executed from top to bottom, and their position in the list can be changed by clicking on them and dragging them to a new position, up or down. Sequences that pass the first filter instance are sent to the second filter instance, etc. Any sequences that do not pass any filter are discarded, so that only sequences that pass all filters are written to the output file. The filter algorithms are widely disparate in their processing requirements. For example, the sequence length filter requires relatively little processing power and is therefore pretty fast. In contrast, the Clustal Omega filter is processor intensive and is, therefore, very slow. To maximize the efficiency of your searches, place the filter order of your methods in order from least intensive processing requirements to most intensive. This will cause the fewest possible sequences to be put through the slowest algorithms in your search. Based on one test we ran, the order of current filters from least processor intensive to most intensive is as follows:
Regex==> SecSig==> Sequence Length==> TM Prediction==> Clustal Omega
This is not a linear list. There is very little difference between the requirements of Regex and SecSig based on our test. There is a very big difference between Clustal Omega and all the others. This is simply an ordering from least intensive to most intensive, A.K.A. fastest to slowest.
Sequence Length Filter. This filter, very simply, filters all sequences in a FASTA file and excludes all but those that are between a user specified minimum and a maximum length. This is useful if the user knows a gene or protein class that she is looking for tends to be about a certain length. When the user double clicks on a filter instance of this type, the following configuration options appear:
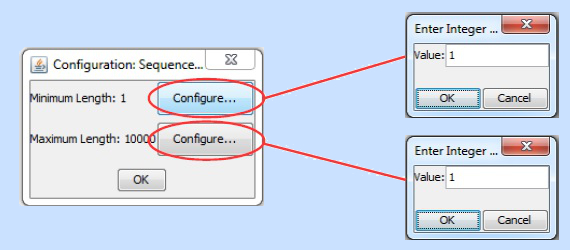
Set the minimum value, press "OK." Set the maximum value, press "OK" and then press "OK" again. The filter instance will now be configured and ready to execute.
Regex Filter. This filter is a regular expressions filter, and is quite powerful. For an introduction on how to use regular expressions see "A Primer on Regex" at the end of this section of the user guide. In the context of genomic and proteomic searches, one can use Regex to find common patterns among large numbers of sequences. For example, one might use Regex to find all sequences containing 3 Arginines that are eight to ten amino acids apart. This example is made up, but perhaps you know this arrangement forms a particular type of active site in some class of enzyme you are interested in. Another example may be all the sequences that have 8 to 20 cysteine residues. This example may produce sequences for proteins that are rich in disulfide bonds. If you are interested in some recent real world applications of regular expressions in genomic research, here are a few examples:
Zelman AK, Dawe A, Berkowitz GA. Identification of cyclic nucleotide gated channels using regular expressions. Methods Mol Biol. 2013;1016:207-24.
Seiler M, Mehrle A, Poustka A, Wiemann S. The 3of5 web application for complex and comprehensive pattern matching in protein sequences. BMC Bioinformatics. 2006 Mar 16;7:144.
Tataru P, Sand A, Hobolth A, Mailund T, Pedersen CN. Algorithms for hidden markov models restricted to occurrences of regular expressions. Biology (Basel). 2013 Nov 8;2(4):1282-95.
When the user double clicks on a filter instance of this type, the following configuration options appear (click on the image to see it in higher resolution):
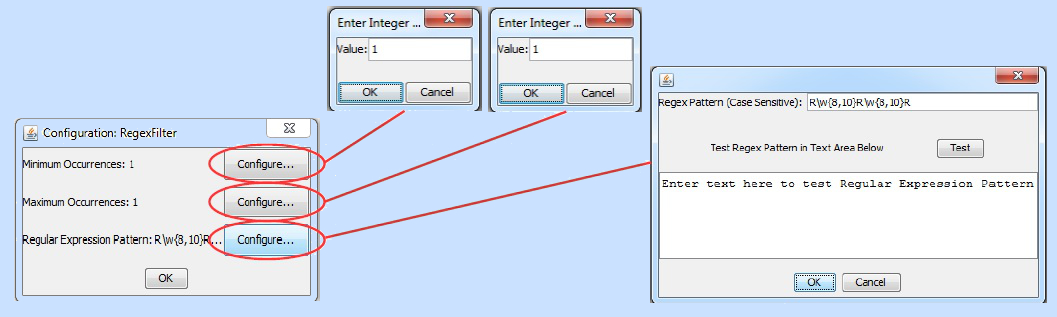
Enter a minimum and maximum number of occurrences in their respective dialogs and press "OK." Then configure your regular expression by entering it into the Regex pattern dialog as shown above. If you wish, you can also test the expression on known matches by entering them into the test text area and pressing the "Test" button. Incidentally, the expression entered in the example here, R\w{8,10}R\w{8,10}R, will match the fictional example we mentioned earlier about an active site that has three arginines each separated by eight to ten amino acids. As entered above, the filter will find sequences that contain one, and only one, match for this pattern due to both the minimum and maximum number of occurrences being set to one.
Transmembrane Prediction Filter (TMPredictionFilter). This filter uses the single sequence version of TMAP to predict transmembrane segments in protein sequences. TMAP is an algorithm that was developed using positional frequencies of amino acids in multiple sequence alignments of homologous proteins containing transmembrane segments. By differentially scoring amino acids of transmembrane and membrane flanking segments vs. non-transmembrane segments and experimenting with different analysis frame sizes they were able to obtain far more accurate predictions than previous in silico methodologies. TMAP is typically used (and was developed to be used) for determining the transmembrane segments in multiple sequence alignments of homologous proteins. It is likely that the single sequence version is less accurate to some extent. However, it is available and multiple sequence alignments would not work with the current code logic of Genomalysis. For a detailed discussion of TMAP development see the publications below:
Persson B, Argos P. Prediction of transmembrane segments in proteins utilising multiple sequence alignments. J Mol Biol. 1994 Mar 25;237(2):182-92.
Persson B, Argos P. Topology prediction of membrane proteins. Protein Sci. 1996 Feb;5(2):363-71.
When the user double clicks on a filter instance of this type, the following configuration options appear:
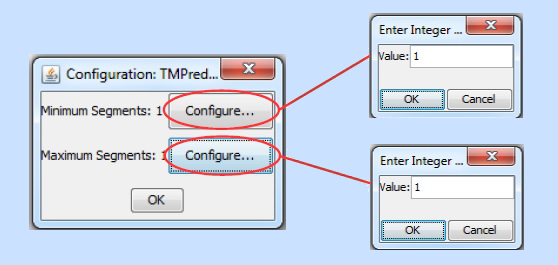
As with the sequence length filter, set the minimum and maximum values, press the respective "OK" buttons, and then press "OK" again. The filter instance will be then be configured and ready for execution.
Secretion Signal Filters (SecSigFilter). These filters use the PrediSi algorithm to check for secretion signals in proteins. The algorithm uses one of three libraries for prediction: one developed for prediction of secretion signals in Gram positive bacteria, one for the same in Gram negative bacteria, and one for eukaryotic secretion signals. Genomalysis has three different available filters for secretion signal prediction, each using a different one of these three libraries. This filter is not set up for configuration and if you double click on an instance you will be informed of this. This filter tests for both a secretion signal and an associated cleavage site: if both are present, then the sequences passes; if either of both are absent, then the sequence fails. For an extensive discussion of the PrediSi algorithm see the reference below:
Hiller K, Grote A, Scheer M, M�nch R, Jahn D. PrediSi: prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W375-9.
Clustal Omega Filter. This filter uses the Clustal Omega algorithm to do sequence alignments of sequences in a FASTA file to a user entered sequence. It then passes or fails each FASTA file sequence based on similarity settings. The user can choose percentages and exact integer values for the number of weak groups, strong groups, and identities that a FASTA sequence must have in common to the user entered sequence in order for it to pass the filter. Clustal Omega is typically used for multiple sequence alignments and has numerous options designed to allow for high quality alignments of very large sets of sequences. Genomalysis uses Clustal to do sequential basic pairwise alignments in rapid succession. This is an atypical usage of Clustal Omega, but it works very well.
For more on Clustal visit their web site: http://www.clustal.org/
When the user double clicks on a filter instance of this type, the following window appears (click on the image to see it in higher resolution):
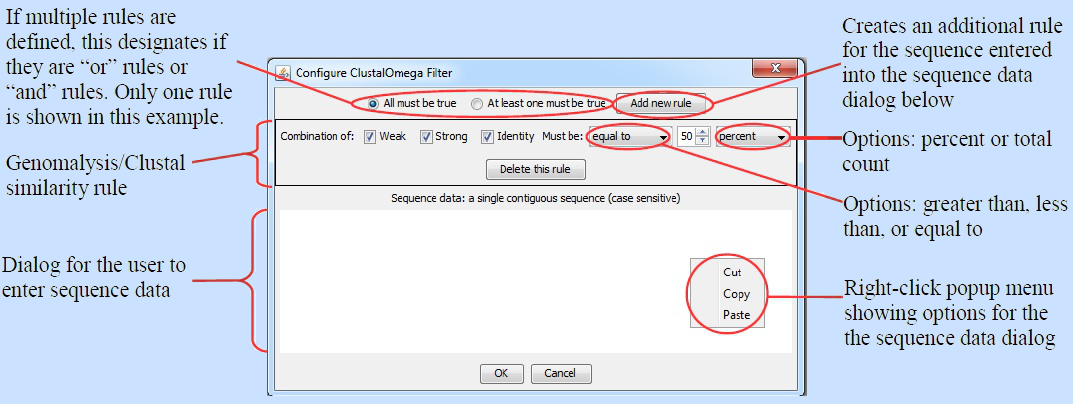
Paste a sequence into the sequence data dialog. Be sure that there are no breaks of any kind in the sequence, for example, a carriage return or a space. If the sequence is not contiguous, then Genomalysis will throw an error when it tries to execute this filter. Set the similarity options in the Genomalysis/Clustal similarity rule. If you need additional rules, then press the "Add New Rule" button and a new rule will appear. All rules added in this way will be applied to FASTA sequences aligned to the sequence in the sequence data dialog of this window. If you need to create FASTA alignment rules for another sequence, then create a second instance of this filter type in the main Genomalysis window and configure it for your additional sequence. When you have chosen all options for all the rules that you need, press the "OK" button. This filter instance will now be configured and ready to be executed.
A Primer on Regex. What is Regex? Regex is a language for expressing patterns. The name "Regex" is from a combination of the words "Regular Expressions." Regex allows you to express patterns with a minimum of typing. These expressions can look very cryptic and scary, but are not particularly difficult to understand if you start with some basics. This primer is geared towards individuals with little or no programming experience and aims to break down the language of Regex into its fundamental parts so that anyone (hypothetically) could understand it.
Why Regex? Regex is useful in situations where you need to detect complicated patterns in text, especially when those patterns can appear multiple times. Regular expressions are portable because they can be used in just about any of the modern programming languages (like Javascript, C#, Java, Ruby, Python, PHP, etc.). There are regular expressions for finding email addresses, URLs, HTML tags or just about any other kind of a pattern that can be expressed in text.
How can we use Regex? Usually, we ask Regex to tell us if it finds a certain pattern in text, and if so, where. Regex can also tell us where the next place is that it sees the pattern. So if we take the statement, "there is a frog on a log" and ask Regex where the word "frog" is located, then it will tell us "position 11." This is the numbered position in the sentence where the word frog begins:
The expression in this case is literally "frog" but we can ask Regex to do more complicated things for us. For example, we can ask Regex to find any word that ends in "og" and tell us where it is. In order to say this to Regex, we break it down as follows: we want a word that has any amount of letters (we don't really care what they are) followed by og. In Regex, the term "\w" signifies a "word letter" (meaning any letter in the alphabet). The "\" tells Regex "this is a special character" and the "w" says "this is a word letter (A - Z)." We can follow the "\w" with an "*" meaning "zero to many" or a "+" meaning "one to many." So to make an expression that says "one to many letters followed by 'og'" we construct the following regular expression: \w+og
Now, when we ask Regex to tell us all of the places where it sees the pattern "\w+og", it tells us "positions 11 and 21."
This is the tip of the iceberg when it comes to the power of regular expressions to perform pattern matching on text. Here, we tell Regex that we want one to many word characters. What if we want exactly one? Just omit the "+" so that we have the following expression: \wog
Or, we might want exactly two. we could do this: \w\wog
But what if we wanted exactly thirteen characters followed by "og"? This situation probably does not come along often, but you never know. we could write "\w\w\w\w\w\w\w\w\w\w\wog" but that is getting a bit ridiculous. Apparently, the creators of regular expressions thought so too, so they created a special notation that you can use to tell Regex exactly how many times you want it to do something. Let's say we want to find all words in some text starting with an "s" and ending with an "n" with exactly 7 letters in between. Here is how we write it: s\w{7}n
This says "s" followed by exactly 7 word characters followed by "n." Pretty cool, huh? But, what if we wanted, say, anywhere between 3 and 7 characters between? We just need to modify our last expression a bit to accommodate this: s\w{3,7}n
When we wrote our expression "s\w{7}n" we were actually telling Regex "s\w{7,7}n" but we can just put "{7}" for short.
Now on to escaped characters. What are escape characters? And why were they escaping in the first place? Worry not, because we actually want some of our characters to escape. In regular expressions, we use the backslash, \, to "escape"certain characters from expressions. This means that when a "w" or an "s" is in front of a backslash, it carries special meaning of some kind for Regex. So far we have only used the "\w" in our expressions to detect word characters. Here is a larger list of commonly used escaped characters:

So now, with our newly acquired expert knowledge of Regex escaped characters, we can track down things like phone numbers and email addresses. There is no escape! Well, you might think that anyway. Before we move on, we have to point out something here: the "." character. Alone, in an expression, it has special meaning: any character. If we were to say "s..p" then it would match with ship, soap, slip, etc. But, if we say "s\.\.p" then literally "s..p" is what will be matched. Why would this be backwards from everything else? We have no idea. But it is worth knowing when you make your expressions. A "." means any character, and a "\." means "." literally. Now, moving on. Let's take a good example of something that we might search for in text: a phone number. Phone numbers can take a variety of forms:
(555)555-5555
(555) 555-5555
555-555-5555
1-555-555-5555
555.555.5555
Let's try to match the "555-555-5555" example. There are 3 digits, followed by a dash, followed by three more digits, followed by a dash, followed by four more digits. So, three digits, \d{3}, followed by a dash, \d{3}-, followed by three more digits, \d{3}-\d{3}, and another dash, \d{3}-\d{3}-, followed by four more digits, \d{3}-\d{3}-\d{4}, and there you have it: \d{3}-\d{3}-\d{4}
This pattern will match phone numbers with the pattern "XXX-XXX-XXXX". But if you look closely, we are actually repeating information again (like in the "\w\w\w\w\" example). Right at the beginning of the pattern, you see "\d{3}-" twice. Is there a way to compact this any more? There is, with logical grouping. In Regex you can logically group different parts of a pattern together with parentheses, and then treat the group as an individual element. So we could rewrite the expression, \d{3}-\d{3}-, as the expression, (\d{3}-){2}, and that would make our final expression as follows: (\d{3}-){2}\d{4}
So parentheses, curly braces (the "{" and "}" characters) and backslashes have special meaning in these patterns: curly braces indicate multiplicity, parentheses logically group things, and backslashes indicate that the next character after it should be treated specially. This is all fine and dandy, until we want to find curly braces, parentheses, or backslashes in text. Imagine a situation where we wanted to find, say, a phone number like "(555) 555-5555." If you tried to use the pattern, (\d{3})\s\d{3}-\d{4}, where the first three digits are surrounded by parentheses, then this would actually match "XXX XXX-XXXX" because Regex thinks that you are trying to logically group the three digits. In order to disillusion our faithful search algorithm, we need to escape our parentheses. Run away! You escape parentheses just like anything else: with a backslash. So to get this expression right, we have to write it like this: \(\d{3}\)\s\d{3}-\d{4}
Now that is what we meant by cryptic looking. If we had shown you something like this in the beginning of this discussion, you probably would have run screaming in the opposite direction. Fortunately for you, we're taking it one step at a time, which should hopefully make it easier to understand. This new pattern is great, but it could be a little better. There is a difference between a "(XXX) XXX-XXXX" phone number and a "(XXX)XXX-XXXX" number, although they look almost exactly the same. One has a space after the parentheses, and one does not. It might not seem like much of a difference, but to a computer the difference is significant. Is there a way to make a pattern that can handle either scenario? Well, we could put a multiplicity constraint in front of the whitespace character to make it zero to one, like this:\(\d{3}\)\s{0,1}\d{3}-\d{4}
That works, but there is a quicker way to say the same thing. Remember the "*" and "+" operators for "zero to many" and "one to many"? There is also an operator for zero to one, the "?" character. So if we rewrite our pattern as, \(\d{3}\)\s?\d{3}-\d{4}, then we have an equivalent expression that says, "exactly three digits surrounded with parentheses followed by an optional space, followed by exactly three digits, one dash, then exactly four digits."
Hopefully, this has helped you get somewhat of a basic understanding of what Regex can do. There are entire books written on the subject, and millions of programs and web sites all over the world that use them. In bioinformatics they can be extremely powerful for finding certain types of sequence elements.
Top of page Previous page Next page